Life Science and Technology News
Quantitative Approach on Understanding How Epigenetic Switches Control Gene Expression
Scientists at Tokyo Institute of Technology decipher how to quantitatively assess the effects of specific epigenetic changes on the rate of transcription by developing a mathematical model. For this, they successfully generated reconstituted chromatin bearing histone modifications in vitro. Their study published in Nucleic Acids Research provides an accurate quantitative approach for understanding how site-specific changes to histone proteins impact the accessibility of chromatin and gene expression levels.
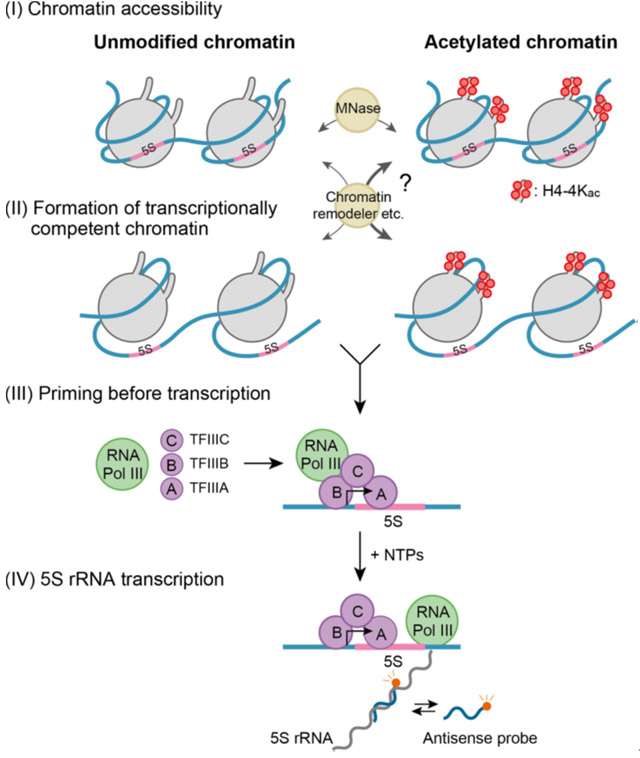
Figure 1. Transcription stages at the chromatin level
Site specific histone acetylation can differentially affect the sequential stages of transcription.
The creation of a single protein is a long and complicated process. Even just the generation of its blueprint itself, known as a coding transcript, from a DNA template involves numerous steps and players. To start with, the DNA is usually found neatly wrapped around proteins called histones to form a nucleosome, the fundamental subunit of a tightly condensed structure called chromatin. The extent of its condensation determines how much of the DNA is "available" for the transcription process. Changes to these histones, such as acetylation, also influence the accessibility of chromatin for gene expression. These "epigenetic" modifications play a crucial role in regulating gene expression. To date, much remains to be explored regarding how these epigenetic effects can be accurately quantified and how site-specific histone modifications can affect gene expression.
To answer these questions, a group of researchers led by Prof. Masahiro Takinoue from Tokyo Institute of Technology, Prof. Kohki Okabe from The University of Tokyo, and Prof. Takashi Umehara from RIKEN Center for Biosystems Dynamics Research, Japan have developed a kinetic model to quantify the contribution of epigenetic changes on transcription rates based on highly quantitative experimental results. Talking about their recent work published in Nucleic Acids Research![]() , Takinoue explains, "The contribution of each modification state of each histone to the sequential steps of chromatin transcription was yet to be quantified because of the difficulty in precise reconstitution of a chromatin template with the epigenetic modification(s) of interest and quantification of RNA transcription from it. Using genetic code expansion and cell free protein synthesis, we synthesized histone H4 containing designed site-specific acetylation(s) and reconstituted a tetra-acetylated nucleosome."
, Takinoue explains, "The contribution of each modification state of each histone to the sequential steps of chromatin transcription was yet to be quantified because of the difficulty in precise reconstitution of a chromatin template with the epigenetic modification(s) of interest and quantification of RNA transcription from it. Using genetic code expansion and cell free protein synthesis, we synthesized histone H4 containing designed site-specific acetylation(s) and reconstituted a tetra-acetylated nucleosome."
Histone modifications target various stages (as described in Figure 1) in the process of transcription at the chromatin level, which could in turn affect the rate of transcription. The first stage, 'chromatin accessibility', involves the opening up of the tightly condensed structure and making it accessible to the transcription machinery. The second stage is the 'formation of transcriptionally competent chromatin', which prepares the chromatin for the binding of transcript synthesizing complexes. The third stage is 'priming before transcription', which aids the assembly of accessory proteins required to begin transcription. The final stage of transcription involves the sequential addition of nucleotides to form the transcript.
To study how site-specific histone modifications, including acetylation, affect the stages of transcription, the researchers created a reconstituted nucleosome carrying two copies of an RNA coding sequence from Xenopus, a frog species, with acetylation on specific sites of the histone proteins. This system simulates the dynamic changes in chromatin within the living cell, which affect transcription. They developed an accurate and highly sensitive fluorescence-based detection system that can measure the minute concentrations of transcripts in real time. On applying their kinetic model (see Figure 2), they found that acetylation at four specific histone sites increases the accessibility of chromatin threefold when compared with chromatin lacking acetylation.
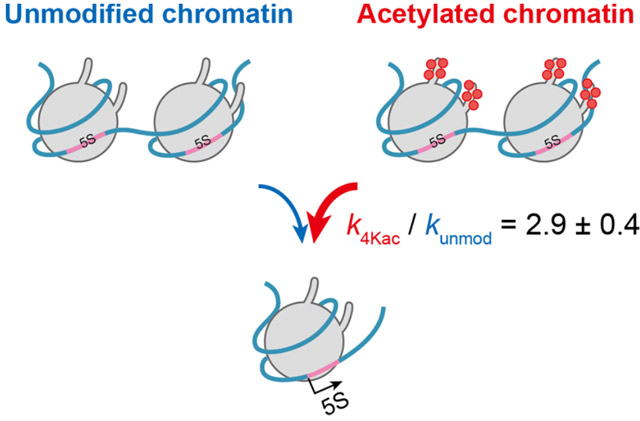
Figure 2. The novel kinetic model
This kinetic model from the study can be applied for quantification of epigenetic changes on transcription rate.
The model's versatility also allows for it to be used for quantifying other epigenetic modifications. Highlighting the potential applications of their study, Takinoue states, "Our mathematically described kinetic model allowed us to determine the rates of chromatin transcription from non-acetylated and tetra-acetylated chromatin templates. Our methodology will be applicable to a wide variety of chromatin-mediated reactions for quantitative understanding of the importance of epigenetic modifications."
- Reference
| Authors : | Masatoshi Wakamori1, Kohki Okabe2,3,*, Kiyoe Ura3,4, Takashi Funatsu2, Masahiro Takinoue3,5,* and Takashi Umehara1,3,* |
|---|---|
| Title of original paper : | Quantification of the effect of site-specific histone acetylation on chromatin transcription rate |
| Journal : | Nucleic Acids Research |
| DOI : | 10.1093/nar/gkaa1050 |
| Affiliations : | 1 Laboratory for Epigenetics Drug Discovery, RIKEN Center for Biosystems Dynamics Research 2 Graduate School of Pharmaceutical Sciences, The University of Tokyo 3 PRESTO, Japan Science and Technology Agency (JST) 4 Graduate School of Science, Chiba University 5 Department of Computer Science, Tokyo Institute of Technology |
- Giving Movement to The Micro: Novel Technique Moves Micro-particles using Electric Fields | Life Science and Technology News
- Designing DNA From Scratch: Engineering the Functions of Micrometer-Sized DNA Droplets | Life Science and Technology News
- Scientists develop DNA microcapsules with built-in ion channels | Life Science and Technology News
- Formation of Artificial Cells with a Skeletal Support Reinforcement to withstand Application Realized | Tokyo Tech News
- Successful use of DNA as a computer in artificial cells - Realizing molecular robots that work in vivo - | Tokyo Tech News
- Microdroplet reactors mimic living systems | Tokyo Tech News
- FY2016 STAR grant recipients selected | Tokyo Tech News
- Takinoue Lab.
- Researcher Profile | Tokyo Tech STAR Search - Masahiro Takinoue
- Department of Computer Science, School of Computing
- Department of Systems and Control Engineering, School of Engineering
- Department of Life Science and Technology, School of Life Science and Technology
- RIKEN Center for Biosystems Dynamics Research
- Graduate School of Pharmaceutical Sciences, Faculty of Pharmaceutical Sciences, The University of Tokyo
- Chiba University
- Latest Research News

School of Computing —Creating the Future Information Society—
Information on School of Computing inaugurated in April 2016
Further Information
Associate Professor Masahiro Takinoue
School of Computing, Tokyo Institute of Technology
Email takinoue@c.titech.ac.jp
Tel +81-45-924-5680
Featured News


![]()