Materials Science and Engineering News
Towards Polymer Informatics: Open-source Library for Creating Polymer Property Databases
Physical property databases for polymers are now much easier to build from molecular dynamics simulations thanks to RadonPy, a new open-source library developed by researchers at the Institute of Statistical Mathematics (ISM) and Tokyo Institute of Technology (Tokyo Tech), Japan. This software automates the simulation and calculation of several relevant properties for a given list of polymers. In turn, this greatly accelerates the generation of large amounts of data to be used in the search for new compounds via materials informatics.

Over the past few years, materials informatics has emerged as a very promising field of study that is revolutionizing how scientists search for new materials. In broad terms, researchers in materials informatics seek to leverage machine learning to predict the properties of materials. However, just like in most applications of machine learning, one first needs to gather huge amounts of data to train the predictive models.
One way to generate this data is through simulations. For example, researchers have already created entire property databases for inorganic crystals by means of molecular simulations. Such databases have been useful in the search for novel compounds for catalytic, electronic, and optical applications, to name a few. Unfortunately, the same cannot be said about polymeric materials. Although this family of materials offers unique characteristics that are essential for many industrial applications, there are currently no large polymer property databases available to propel materials informatics forward.
In an effort to address this problem, a team of scientists from the Institute of Statistical Mathematics (ISM) and Tokyo Institute of Technology (Tokyo Tech), Japan, created RadonPy, an open-source Python library designed to streamline the process of conducting thousands of molecular dynamics simulations for polymers. This study, which was led by Professor Junko Morikawa, was published in npj Computational Materials![]() .
.
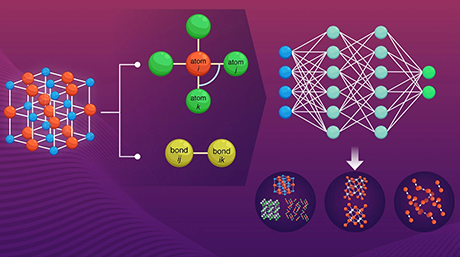
To use RadonPy, one simply needs to input the repeating unit of a desired target polymer using a standardized notation and provide a few parameters, such as the degree of polymerization and number of polymer chains to consider in the simulation. The program takes the wheel from there and performs all the necessary steps to conduct all-atom classical molecular dynamics simulations, repeating any that do not converge or fail to reach an equilibrium state. Finally, the algorithm performs additional calculations to determine an assortment of physical properties for the polymer, such as its thermal conductivity, refractive index, density, and specific heat capacity.
One of the most attractive features of RadonPy is that it was optimized to be run on highly parallel computers, such as supercomputers. This means that one can provide a long list of target polymers as an input and the program will efficiently split the tasks between the processing nodes. The researchers tested this first release of RadonPy by calculating 15 properties of over 1,000 amorphous polymers and validating these properties against available experimental data. By focusing specifically on the thermal conductivity of these polymers, they obtained some interesting results. Prof. Yoshida remarks, "We computationally identified eight amorphous polymers with extremely high thermal conductivities, six of which were yet unreported. Moreover, a feature implemented in RadonPy enabled us to determine the underlying mechanisms behind these high thermal conductivity values."
Overall, the research team has high hopes for RadonPy and will continue to promote its further development. "Similar to the advancement of materials informatics since the advent of computational property databases for inorganic crystals, database construction using RadonPy will promote the development of polymer informatics," comments Prof. Morikawa. Hopefully, this software will make it so polymer databases are more readily available, ultimately leading to the discovery of useful materials.
- Reference
| Authors : | Yoshihiro Hayashi1,2, Junichiro Shiomi1,2,3, Junko Morikawa1,4, and Ryo Yoshida1,5,6 |
|---|---|
| Title : | RadonPy: automated physical property calculation using all-atom classical molecular dynamics simulations for polymer informatics |
| Journal : | npj Computational Materials |
| DOI : | 10.1038/s41524-022-00906-4 |
| Affiliations : | 1The Institute of Statistical Mathematics (ISM), Research Organization of Information and Systems 2Department of Mechanical Engineering, The University of Tokyo 3Institute of Engineering Innovation, The University of Tokyo 4Department of Materials Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology 5Research and Services Division of Materials Data and Integrated System (MaDIS), National Institute for Materials Science (NIMS) 6Department of Statistical Science, School of Multidisciplinary Science, The Graduate University of Advanced Studies (SOKENDAI) |
|
* Corresponding authors' emails: yhayashi@ism.ac.jp; yoshidar@ism.ac.jp |
|
- Successful application of machine learning in the discovery of new polymers | Tokyo Tech News
- A micro-thermometer to record tiny temperature changes | Tokyo Tech News
- The Joint instruction program with RWTH Aachen University | Tokyo Tech News
- Morikawa Lab
- Junko Morikawa | Researcher Finder - Tokyo Tech STAR Search
- RadonPy project RadonPy|GitHub
- Polymer Database (PoLyInfo) | National Institute for Materials Science
- Human Centered Science and Biomedical Engineering Graduate Major|Education|Department of Materials Science and Engineering, School of Materials and Chemical Technology
- Materials Science and Engineering Graduate Major|Education|Department of Materials Science and Engineering, School of Materials and Chemical Technology
- Materials Science and Engineering Undergraduate Major|Education|Department of Materials Science and Engineering, School of Materials and Chemical Technology
- National Institute for Materials Science (NIMS)
- The Institute of Statistical Mathematics
- Research Center for Computational Science
- The University of Tokyo
- Latest Research News

School of Materials and Chemical Technology
—Encompassing the Disciplines of Science—
Information on School of Materials and Chemical Technology inaugurated in April 2016
Further Information
Professor Junko Morikawa
School of Materials and Chemical Technology, Tokyo Institute of Technology
Email : morikawa.j.aa@m.titech.ac.jp
Tel +81-3-5734-2497
Featured News

![]()